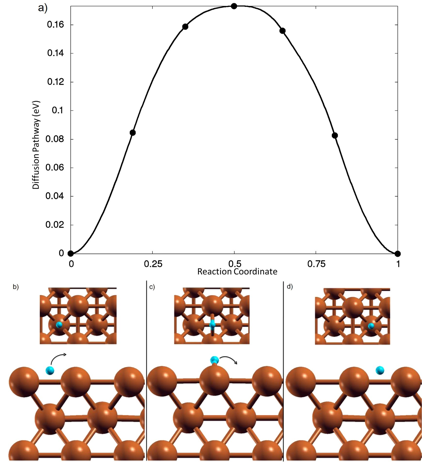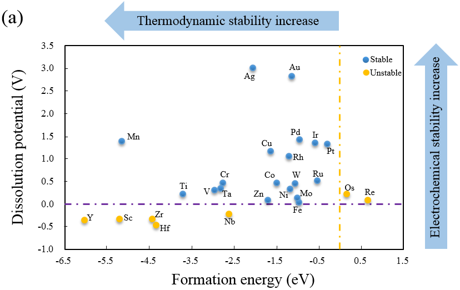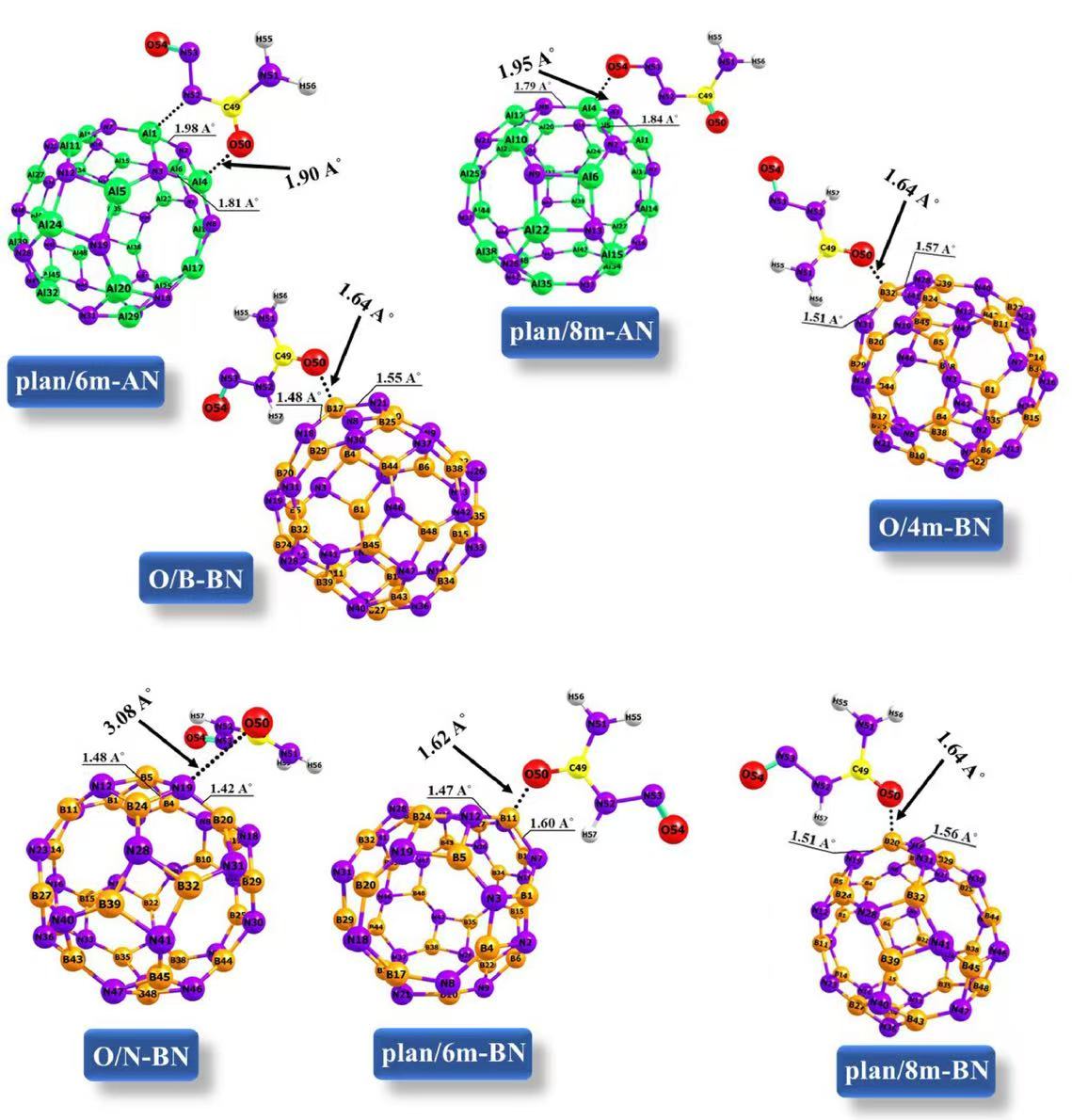
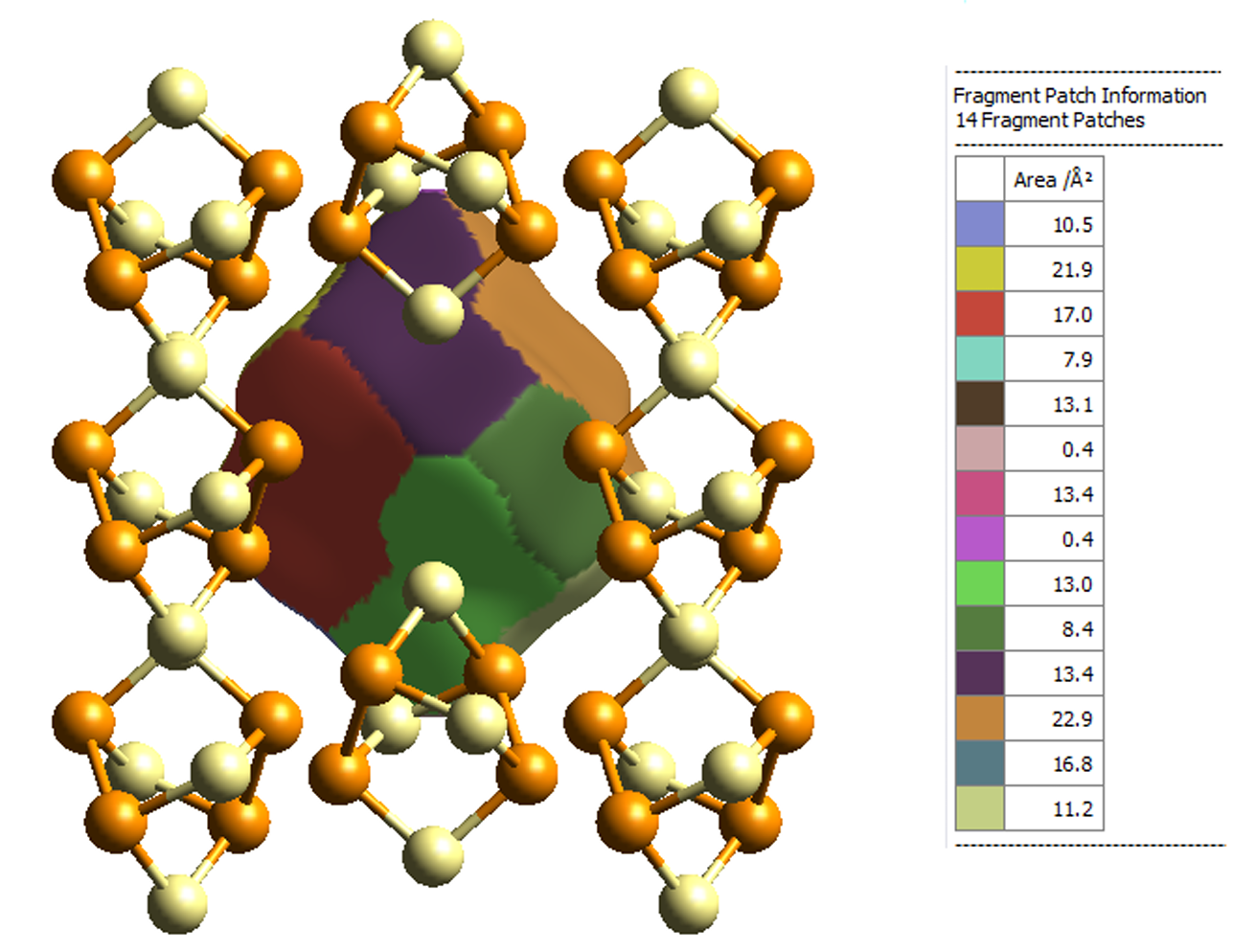
在计算化学和材料科学中,Hirshfeld 电荷、Mulliken 电荷和 Bader 电荷是三种常用的电荷分析方法,用于量化分子或固体中原子的电荷分布。它们的理论基础、计算方法和适用场景各有差异。
基于分子轨道理论(MO 理论),通过原子轨道在分子轨道中的贡献来划分电荷。
核心假设:分子轨道由原子轨道线性组合而成(LCAO-MO),电荷按原子轨道在分子轨道中的系数平方分配。
基于独立原子模型(Independent Atom Model, IAM),通过将分子中的电子密度与自由原子的电子密度对比,划分原子间的电荷。
核心思想:每个原子在分子中的电荷等于其自由原子的电子数减去分子中该原子区域内的电子数。
基于量子化学中的原子在分子(Atoms in Molecules, AIM)理论,由 Richard Bader 提出,通过电子密度的拓扑分析划分原子区域。
核心概念:以电子密度梯度的临界点(如键临界点、核临界点)定义原子间的边界,每个原子区域为Voronoi 型分区,满足电荷守恒。
| 特征 | Mulliken 电荷 | Hirshfeld 电荷 | Bader 电荷 |
|---|---|---|---|
| 理论基础 | 分子轨道理论(均摊法) | 独立原子模型(电子密度权重) | AIM 理论(电子密度拓扑) |
| 电子密度依赖性 | 间接(通过轨道系数) | 直接(对比自由原子密度) | 直接(基于实际密度分布) |
| 基组敏感性 | 高 | 中 | 低 |
| 适用体系 | 小分子、定性分析 | 有机分子、极性化合物 | 强相互作用体系、固体 |
| 结果合理性 | 定性趋势合理,定量误差大 | 定量更合理(尤其极性键) | 最接近实际电荷分布 |
| 计算复杂度 | 低(软件默认支持) | 中(需自由原子密度) | 高(需拓扑分析) |
实际应用中,常结合多种方法交叉验证,例如同时计算 Mulliken 和 Bader 电荷,对比分析体系的电荷离域程度或计算方法的可靠性。





长按屏幕识别二维码
打开手机扫描二维码