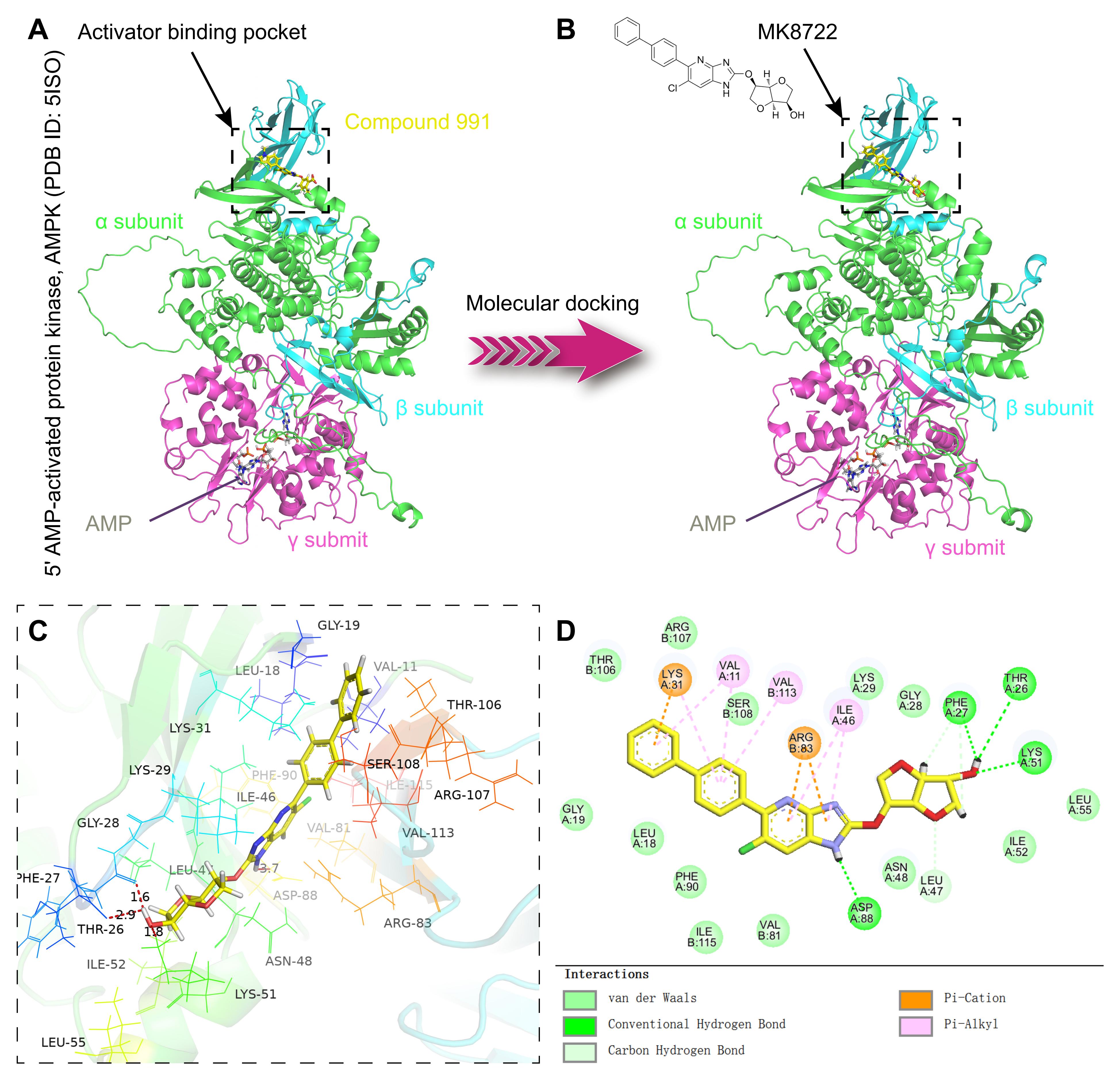
分子对接(Molecular Docking) 是一种通过计算模拟小分子(如药物候选化合物)与靶标分子(如蛋白质、核酸)之间的相互作用,预测两者结合模式和亲和力的技术。其核心目标是通过模拟分子间的空间匹配和能量互补,筛选出潜在的高活性化合物,广泛应用于药物设计、虚拟筛选和分子识别研究中。
基本原理与核心概念
1. 分子识别的基础
- 空间匹配:小分子的官能团、原子位置需与靶标活性位点的空腔形状、氨基酸残基分布互补(如 “钥匙 - 锁” 模型)。
- 能量互补:分子间通过非共价键(氢键、范德华力、疏水作用、静电作用等)相互作用,形成稳定复合物,体系能量降低。
2. 关键参数
- 结合能(Binding Energy):衡量分子间结合的稳定性,通常通过热力学公式估算(如自由能变化 \(\Delta G\)),负值越大表示结合越紧密。
- 对接分数(Docking Score):算法根据空间匹配和能量参数生成的综合评分,用于排序候选化合物的结合潜力。
分子对接的流程
1. 准备靶标分子(受体,Receptor)
- 结构获取:从蛋白质数据库(PDB)下载靶标蛋白的三维结构,需去除无关配体、水分子,修复缺失氨基酸(如 Loop 区)。
- 活性位点定义:指定对接区域(如酶的催化位点、受体的配体结合口袋),可通过实验数据(如突变分析)或软件自动识别(如 AutoDock 中的 “Grid Box”)。
2. 准备配体分子(Ligand)
- 结构构建:通过分子编辑器(如 ChemDraw)绘制小分子结构,或从数据库(如 ZINC、PubChem)获取。
- 构象生成:配体可能存在多个可旋转键,需生成其低能构象集合(如使用 Open Babel、MOPAC),以覆盖不同结合取向。
3. 对接计算
- 选择对接算法:
- 刚性对接(Rigid Docking):受体和配体均不发生构象变化,适用于快速筛选(如 AutoDock Vina)。
- 半柔性对接(Semi-flexible Docking):配体可旋转部分单键,受体保持刚性(最常用,如 AutoDock、Glide)。
- 柔性对接(Flexible Docking):受体和配体均允许构象变化(计算成本高,如分子动力学模拟结合对接)。
- 搜索空间采样:通过算法(如遗传算法、分子动力学、蒙特卡洛模拟)探索配体在受体活性位点的所有可能取向,寻找能量最低构象。
常用软件与工具
软件 | 特点 | 适用场景 |
|---|
AutoDock | 开源,支持半柔性对接,经典力场计算,适合学术研究 | 大规模虚拟筛选 |
AutoDock Vina | 速度快,对接精度高,用户界面友好(基于 AutoDock 改进) | 快速先导化合物筛选 |
Glide (Schrödinger) | 商业软件,高精度对接,考虑受体柔性和溶剂效应 | 药物研发后期精细化优化 |
DOCK | 基于形状匹配的刚性对接,适合初步筛选 | 高通量虚拟筛选 |
GOLD | 采用遗传算法,灵活处理配体构象,可自定义评分函数 | 复杂结合模式分析 |
Surflex-Dock | 基于受体表面特征匹配,适合片段对接(Fragment Docking) | 苗头化合物发现 |
应用领域
1. 药物设计与发现
- 虚拟筛选(Virtual Screening):从数百万个化合物库中快速筛选与靶标结合的潜在药物(如新冠病毒主蛋白酶 M<sup>pro</sup>的抑制剂筛选)。
- 先导化合物优化:通过对接分析优化化合物结构(如调整取代基位置增强氢键相互作用),提高亲和力和选择性。
2. 生物分子相互作用研究
- 解析配体 - 受体、蛋白质 - 蛋白质、蛋白质 - DNA 等复合物的结合机制(如转录因子与 DNA 的识别模式)。
- 预测小分子的脱靶效应(Off-target Effects),评估药物安全性。
3. 材料科学与催化
- 模拟小分子在催化剂表面的吸附行为(如 CO<sub>2</sub>在金属有机框架(MOF)中的吸附模式)。
局限性与挑战
1.模型简化的局限性
- 对接过程通常假设受体为刚性结构,忽略蛋白质动态构象变化(如诱导契合效应),可能导致假阴性结果。
- 部分算法未显式考虑溶剂分子和离子的影响,需通过隐式模型近似(如泊松 - 玻尔兹曼方程)。
2.评分函数的不足
- 对接分数与实际结合能的相关性有限,可能漏筛高活性化合物(如熵效应难以准确计算)。
- 不同软件的评分函数差异大,需结合实验数据交叉验证。
3.计算资源需求
- 柔性对接和大规模筛选需高性能计算(HPC)支持,限制了中小实验室的应用。
发展趋势
- 结合人工智能(AI):如使用深度学习预测结合亲和力(如 AlphaFold2 预测复合物结构),或通过强化学习优化化合物结构。
- 多尺度模拟整合:对接与分子动力学(MD)、量子化学计算结合,更真实模拟分子动态行为。
- 片段对接(Fragment Docking):以小片段为起点,逐步组装高亲和力化合物,提高筛选效率。
总结
分子对接是连接计算模拟与实验研究的关键桥梁,其高效性和低成本特性使其成为现代药物研发的核心工具。尽管存在模型简化和计算瓶颈,随着算法改进和算力提升,分子对接正朝着更精准、更智能的方向发展,未来将在个性化医疗、新型靶点开发等领域发挥更重要作用。